- CHIMIE THÉORIQUE
- CHIMIE THÉORIQUELa chimie théorique est une discipline neuve, apparue vers 1930. Se développant lentement jusqu’à la Seconde Guerre mondiale, puis plus rapidement dans les années cinquante, elle a connu un essor important depuis 1960 grâce au progrès des ordinateurs. Ainsi, dans de nombreuses universités scientifiques françaises et étrangères, cette discipline a acquis droit de cité au même titre que celles beaucoup plus anciennes et traditionnelles de la chimie: chimie physique, chimie minérale, chimie organique.Son objectif est l’application de la mécanique quantique ou ondulatoire aux problèmes de la chimie. Il s’agit aussi bien de calculer une propriété chimique, à partir des équations de base de cette mécanique, que de fournir les concepts nécessaires à la compréhension des phénomènes. Dans la grande majorité des cas, les principes et les équations de base interviennent pour permettre au théoricien de déterminer le comportement des électrons dans les molécules. Ce comportement détermine à son tour la géométrie et la structure des molécules ainsi que leur aptitude à entrer en réaction et les changements et déformations qu’elles subissent lorsqu’elles réagissent. Il faut cependant, pour décrire les réactions et avoir une vue d’ensemble des phénomènes, faire également appel à certains concepts traditionnels de mécanique classique.Les outils de travail du chimiste théoricien sont donc la plume et le papier, et surtout les machines à calculer de toutes dimensions, depuis les micro-ordinateurs personnels jusqu’aux plus puissants calculateurs (I.B.M. 3090 ou Cray-2). En effet, l’équation fondamentale de la mécanique quantique, qui contrôle le comportement des électrons et des noyaux dans une molécule, ne peut être résolue que par des méthodes numériques approchées. Ces dernières font appel au calcul de millions d’intégrales compliquées pour une seule molécule. Il faut alors utiliser des programmes spécialisés qui font partie de l’arsenal de tous les laboratoires théoriques. De nos jours, il n’est pas rare de voir obtenues par ordinateur la géométrie optimisée de molécules aussi compliquées que des polypeptides, des complexes métalliques, des fragments d’enzymes ou d’acides nucléiques, ainsi que de molécules entourées de leur solvant ou même de l’eau à l’interface de deux couches lipidiques. Cependant, l’interprétation même des phénomènes, qu’ils soient observés par des équipes expérimentales ou qu’ils résultent de puissants calculs, est en dernier ressort un problème purement intellectuel où la logique, la déduction et la synthèse sont requises.1. HistoriqueLa chimie théorique est, à ses débuts, un exercice passager pour physiciens. Après la découverte de la dualité onde-corpuscule de l’électron par de Broglie en 1924 et de l’équation de Schrödinger en 1926, les savants de l’époque souhaitent vérifier la validité et l’applicabilité de ces nouveaux principes. En 1927, Heitler et London en Allemagne calculent l’énergie de la molécule d’hydrogène H2. Dès 1931, Hückel parvient à traiter une molécule aussi complexe que le benzène, grâce à une méthode utilisant des ondes électroniques ou orbitales recouvrant le squelette des atomes de carbone. En 1933, Mulliken, aux États-Unis, étend cette méthode des orbitales moléculaires à de très nombreuses molécules et jette les fondements de la spectroscopie moderne en interprétant les spectres d’absorption des molécules dans l’ultraviolet à l’aide de ces orbitales. La transition d’un électron entre deux orbitales accompagne l’absorption d’un photon par la molécule. La parution d’Introduction to Quantum Mechanics de Pauling et E. B. Wilson, en 1937, et de The Nature of the Chemical Bond par Pauling, en 1939, marque la reconnaissance définitive d’une nouvelle discipline.C’est sans doute par Lennard-Jones qu’est fondé le premier «laboratoire» de chimie théorique, à Cambridge (G.-B.). De 1933 jusqu’à la mort du fondateur, en 1953, les bureaux sont installés dans le grenier du Cavendish Laboratory, Free School Lane. Son successeur, Longuet-Higgins, accueille simultanément des théoriciens devenus depuis fameux, comme Boys, Griffith, Orgel, Pople, Roothaan. À Oxford, dans l’université concurrente, Coulson, ancien élève de Lennard-Jones, crée un laboratoire semblable.En 1943, Raymond Daudel fonde un centre de chimie théorique à Paris, qui acquiert rapidement une réputation mondiale. En 1948 s’y tient le premier grand colloque international d’après-guerre, avec la participation de Mulliken, Pauling, Coulson, Longuet-Higgins, entre autres. Deux importants laboratoires parisiens sont ensuite créés: le Centre de mécanique ondulatoire appliquée, dirigé par Raymond Daudel, et le laboratoire de chimie quantique de l’Institut de biologie physico-chimique, dirigé par Bernard Pullman. Depuis 1965, d’autres laboratoires ont pris naissance sur tout le territoire français: Bordeaux, Grenoble, Lyon, Marseille, Nancy, Orsay, Paris, Pau, Rennes, Strasbourg, Toulouse...Il y a actuellement des laboratoires de chimie théorique dans le monde entier. Ils sont nombreux aux États-Unis, surtout dans les universités de haut niveau (Harvard, Calif. Techn., Chicago, Cornell) ou encore dans des centres bien équipés en ordinateurs (I.B.M. à San Jose, Bell Labs).2. Les bases de la chimie théoriqueL’équation de SchrödingerL’équation fondamentale de la chimie théorique est l’équation de Schrödinger:
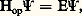 qui signifie essentiellement: «L’opération de l’opérateur hamiltonien H sur la fonction d’onde 切, fonction des coordonnées de toutes les particules (noyaux et électrons), donne la même fonction 切 multipliée par un nombre E. Le nombre E est l’énergie du système. L’équation admet généralement un ensemble de solutions E, correspondant aux différentes énergies possibles. On a donc un ensemble de niveaux d’énergie discrets pour la molécule, c’est-à-dire discontinus, ce qui est une caractéristique primordiale de la mécanique quantique (en mécanique classique, l’énergie d’un système peut varier continûment).L’opérateur hamiltonien Hop contient à la fois un terme différentiel, assimilable à l’énergie cinétique des particules, et un terme multiplicatif, assimilable à leur énergie potentielle. C’est la présence simultanée de ces deux termes de caractère mathématique très différent qui rend la résolution de l’équation si difficile.En pratique, une première simplification est apportée en séparant noyaux et électrons. Comme le noyau le plus petit, le proton, est déjà 1 860 fois plus lourd qu’un électron, on suppose que les électrons se déplacent beaucoup plus rapidement que les noyaux. Ainsi, du point de vue d’un électron en mouvement, les noyaux sont essentiellement fixes. C’est l’approximation de Born-Oppenheimer: les noyaux n’ont pas eu le temps de bouger alors que déjà les électrons ont accompli de nombreuses trajectoires.Il reste que, même réduite aux seuls électrons, l’équation de Schrödinger est impossible à résoudre analytiquement, sauf pour le cas simple d’un électron (atome hydrogène H ou ions hélium He +, lithium Li ++...). Il faut donc trouver des méthodes approchées; les deux méthodes utilisées communément correspondent chacune à un point de vue différent pour la molécule.La méthode des liaisons de valenceL’idée de la méthode des liaisons de valence est que la meilleure fonction d’onde pour une molécule est celle qui superpose les fonctions partielles, correspondant chacune à un mode satisfaisant de liaison pour la molécule. Ainsi, pour la molécule d’hydrogène H2, le chimiste peut intuitivement envisager deux modes possibles de liaison pour les deux électrons (fig. 1 a).Le premier mode correspond à un partage des deux électrons, provenant de chaque atome, entre les deux atomes, pour former une liaison «covalente». Dans le second mode, l’un des atomes accapare les deux électrons et porte ainsi une charge électrique négative; l’autre atome, dépouillé de son électron, porte une charge électrique positive. L’attraction coulombienne entre ces deux charges crée une liaison «ionique». La méthode des liaisons de valence superpose, pour construire la fonction d’onde globale 切, les fonctions d’onde correspondant à ces deux types de liaison.De la même manière, dans la molécule de benzène, les atomes de carbone – chacun pouvant former quatre liaisons avec les atomes voisins – ont le loisir d’organiser leurs liaisons de deux manières (fig. 2 a). La fonction d’onde 切 correcte est une superposition des fonctions d’onde correspondant à ces deux structures. Chimiquement, cette construction est d’autant plus satisfaisante qu’elle s’accorde avec la notion de résonance introduite par Pauling, dans laquelle la molécule de benzène est supposée en quelque sorte alterner entre ces deux structures (ou en tout état de cause emprunter ses caractéristiques chimiques simultanément à ces deux structures). On caractérise cette résonance par une flèche à double pointe.L’intérêt principal de cette méthode est de transcrire en termes quantiques le langage quotidien des chimistes qui représentent la molécule par une ou plusieurs structures de Lewis où les liaisons entre atomes proviennent de la mise en commun d’une paire d’électrons. Dans la méthode de la liaison de valence, on associe une fonction d’onde précise à chaque structure de Lewis. Malheureusement, cette simplicité conceptuelle s’accompagne de grandes difficultés numériques qui viennent seulement d’être résolues. C’est la raison pour laquelle la théorie de la liaison de valence a été nettement moins développée que la théorie des orbitales moléculaires.La méthode des orbitales moléculairesL’autre grande méthode de la chimie théorique consiste à attribuer à chaque électron de la molécule une onde (orbitale moléculaire ) recouvrant toute la molécule. Chaque molécule contient un certain nombre d’orbitales moléculaires ou ondes de type monoélectronique, obtenues comme solutions d’une équation de Schrödinger simplifiée, dite équation de Hartree-Fock. Dans cette équation, on obtient les orbitales moléculaires à l’aide d’une approche autocohérente.Ainsi, pour la molécule d’hydrogène H2, on peut construire, à partir des orbitales atomiques 1s A et 1s B des deux atomes d’hydrogène H, deux orbitales moléculaires, 祥 et 祥 (fig. 1 b). L’une de ces orbitales, 祥, a une amplitude forte entre les deux noyaux. Il lui correspond une forte densité électronique, mesurée par | 祥|2, dans la région de la liaison, ce qui tend à rapprocher les noyaux. Pour cette raison, on l’appelle orbitale moléculaire liante . L’autre orbitale moléculaire 祥 a son amplitude partagée en deux régions de signe opposé. L’un des volumes, qui symbolise l’extension de l’onde, est en gris pour indiquer le changement de signe. Sa densité dans la région internucléaire est très faible, et les noyaux tendent à se repousser; c’est une orbitale antiliante . L’orbitale liante 祥 a une énergie plus basse que celle des orbitales atomiques des atomes séparés, alors que l’orbitale antiliante a une énergie plus élevée.Dans le modèle des orbitales moléculaires, les électrons peuvent occuper par paires les orbitales de plus basse énergie. Dans ces paires, les deux électrons ont leur spin opposé, le spin étant un moment cinétique de rotation intrinsèque de l’électron, conçu comme une «toupie» autour de son axe propre. La limitation au nombre de deux électrons par orbitale est une conséquence du principe d’exclusion de Pauli, qui impose des restrictions de symétrie sur la nature de la fonction d’onde globale 切. Ainsi, les deux électrons de la molécule d’hydrogène occupent l’orbitale liante. On schématise le gain d’énergie dans la molécule par un diagramme où les électrons sont symbolisés par des flèches indiquant la direction de leur spin (fig. 1 c).Dans les hydrocarbures conjugués, comme le benzène, un rôle particulier est joué par les orbitales moléculaires 神 bâties sur les orbitales atomiques 2p z des atomes de carbone (fig. 2 b). Les paires d’électrons qui occupent des orbitales moléculaires de ce type sont délocalisées sur toute la molécule. Il y a trois paires, qui se répartissent uniformément sur les six liaisons carbone-carbone. La description orbitale moléculaire est donc intermédiaire entre les deux structures de résonance (voir méthode précédente). Dans l’une ou l’autre méthode, on aboutit correctement à une molécule de benzène symétrique avec six liaisons carbone-carbone d’égale longueur.3. Quelques découvertes importantes de la chimie théoriqueSurfaces de potentiel et «état de transition»En partant de l’approximation de Born-Oppenheimer, le chimiste théoricien peut calculer l’énergie d’une molécule pour n’importe quelle disposition relative des noyaux. Ainsi, pour une molécule diatomique, il peut évaluer l’énergie en fonction de la distance entre les deux noyaux. Il obtient une courbe appelée courbe de potentiel (fig. 3 a). Cette courbe de potentiel, comme ses formes plus compliquées appelées surfaces de potentiel lorsque le nombre d’atomes est plus grand, représente en quelque sorte la géographie «énergétique» du terrain (vallées, montagnes) que doit emprunter la molécule quand elle se déforme (fig. 3 b).Pour une molécule diatomique, la courbe de potentiel a un puits dont le fond correspond à la position d’équilibre de la molécule, entourée d’un côté par un mur brusque (noyaux trop rapprochés) et de l’autre par une montée plus douce. Cette pente douce correspond à l’élongation progressive de la liaison et à la dissociation de la molécule. Cette forme de courbe est appelée courbe de Morse .Pour réagir, une molécule doit quitter la vallée correspondant à sa géométrie initiale de réactif, pour trouver une autre vallée, correspondant à sa nouvelle forme en tant que produit de la réaction. Si plusieurs molécules entrent en réaction, la situation topologique reste la même. La ou les molécules doivent donc franchir un col pour réagir (fig. 3 c). Elles choisissent le col le plus bas qui puisse relier les deux vallées. L’existence et le rôle de ce col furent soupçonnés par Kohnstamm et Scheffer aux Pays-Bas en 1910, puis découverts et étudiés en premier par Evans et Polanyi, et séparément par Eyring en 1935. Ce dernier l’appela «état de transition». De nos jours, les théoriciens savent calculer la géométrie détaillée de ces états de transition, qui sont en fait des structures ressemblant à des molécules très déformées, où certaines liaisons sont étirées ou même coupées.Le paramagnétisme de la molécule d’oxygèneEn très grande majorité, les molécules sont neutres au point de vue magnétique. Placées dans un champ magnétique externe, elles développent seulement un petit moment magnétique induit qui s’oppose à la direction du champ. Cependant, certaines molécules se comportent comme de véritables petits aimants: elles possèdent un moment magnétique permanent. C’est le cas de la molécule d’oxygène 2: un récipient d’oxygène liquide placé en équilibre sur le plateau d’une balance fait pencher celle-ci si on approche un aimant. Cette aimantation permanente des molécules d’oxygène, dont l’effet au niveau microscopique est de renforcer le champ externe, s’appelle paramagnétisme . Un des premiers grands succès de la méthode des orbitales moléculaires a été l’explication de ce paramagnétisme.La molécule 2 contient 16 électrons, chacun des atomes d’oxygène en fournissant 8. Ces 16 électrons entrent dans les orbitales moléculaires liantes, en commençant par les plus basses. Si on laisse de côté les deux électrons très profonds de type 1s de la couche interne de chaque atome (quatre en tout), et les électrons demi-profonds de type 2s , aussi au nombre de deux par atome (quatre en tout), il reste huit électrons à placer. À partir des orbitales atomiques 2p x , 2p y , 2p z des deux atomes, on peut former trois orbitales moléculaires liantes et trois orbitales antiliantes. Six électrons entrent dans les orbitales moléculaires liantes (fig. 4). Il reste encore deux électrons; bon gré mal gré, ceux-ci entrent dans les orbitales antiliantes, dont il se trouve que les deux plus basses ont la même énergie. D’après la règle de Hund, les deux électrons doivent occuper chacun une orbitale , en gardant leurs spins dans le même sens. Comme à chaque spin d’électron est associé un petit moment magnétique, la molécule possède au total un moment magnétique correspondant à deux spins d’électron. C’est ce qui est vérifié expérimentalement.L’aromaticité et la règle de HückelLes molécules telles que benzène, naphthalène ou anthracène font partie de la famille des molécules aromatiques (cf. BENZOÏDES). Terme utilisé originellement pour caractériser l’odeur forte de ces molécules, l’«aromaticité» a depuis pris le sens d’une stabilité exceptionnelle de ces molécules hydrocarbonées cycliques ou polycycliques par rapport aux molécules analogues à chaînes ouvertes [cf. AROMATICITÉ].Cette stabilité s’explique facilement par la méthode des orbitales moléculaires. En effet, pour un anneau d’atomes de carbone, les orbitales moléculaires 神 se rangent en énergie croissante de la façon suivante: une orbitale très basse, suivie par des paires d’orbitales d’énergie semblable, enfin une orbitale très haute (fig. 5).Ainsi, pour un anneau avec 4n + 2 atomes (6, 10, 14...), il y a 2n + 1 orbitales liantes, suivies de 2n + 1 orbitales antiliantes. Les 4n + 2 électrons de type 神 remplissent de façon satisfaisante toutes les orbitales moléculaires liantes, et forment une couche «fermée» semblable à celle des atomes les plus inertes (hélium, He; néon, Ne; argon, Ar). Par contre, pour un anneau à 4n atomes (4, 8, 12...), la paire d’orbitales liantes et la paire d’orbitales antiliantes les plus centrales sont remplacées par une paire d’orbitales de caractère «non liant»: leur occupation par les électrons n’aide pas à la formation des liaisons carbone-carbone, mais ne s’y oppose pas violemment non plus. Deux électrons 神 (sur les 4n ) occupent chacun une de ces orbitales non liantes. La couche électronique est «ouverte» et la molécule beaucoup plus réactive. Les anneaux à 6, 10 ou 14 électrons doivent donc être particulièrement stables: c’est la règle de Hückel. Si l’on considère les périmètres des molécules de benzène, naphthalène et anthracène, ils ont précisément 4n + 2 atomes, et aussi 4n + 2 électrons 神. Les liaisons transannulaires du naphthalène et de l’anthracène ne modifient que très peu la grande stabilité structurelle due à ces périmètres.Structure électronique des composés inorganiquesLa théorie des orbitales moléculaires a également joué un rôle de premier plan en chimie organométallique, c’est-à-dire dans l’étude des composés possédant simultanément des atomes de métal et des molécules organiques. La structure du ferrocène, synthétisé en 1951, est un des premiers exemples dans ce domaine: la description de la liaison chimique dans cette molécule est restée longtemps obscure car il est impossible d’écrire une structure de Lewis covalente pour ce composé. La théorie des orbitales moléculaires apporte ici une description élégante de la liaison: il se forme plusieurs orbitales moléculaires liantes entre les orbitales d du métal et les orbitales moléculaires 神 de chacun des cycles pentadiényle (fig. 6). À nouveau, les électrons sont accueillis par paires dans chacune de ces orbitales moléculaires, ce qui confère une grande stabilité à cet édifice.Un autre exemple, où la théorie a joué un rôle primordial, est celui de la découverte du cyclobutadiène, anneau instable à quatre électrons. La synthèse directe de ce composé par les méthodes traditionnelles de la chimie organique est restée longtemps infructueuse. Ce sont deux théoriciens anglais, Orgel et Longuet-Higgins, qui, à partir de l’analyse de la structure électronique de ce composé, proposèrent une nouvelle voie de synthèse utilisant des dérivés métalliques. C’est effectivement par cette voie que la première synthèse du cyclobutadiène fut accomplie deux années plus tard.Enfin, la chimie théorique a aussi su expliquer, sans malheureusement avoir pu la prédire, l’existence surprenante des composés de gaz rares, tels que le difluorure ou l’hexafluorure de xénon (XeF2 ou Xe6). Elle a contribué ainsi à faire disparaître le mythe de l’inertie chimique des atomes tels que le xénon ou le krypton.Le rôle de la symétrie dans le contrôle des réactions chimiquesC’est principalement aux savants américains Woodward (prix Nobel 1965) et Hoffmann (prix Nobel 1981) ainsi qu’au chimiste japonais Fukui (prix Nobel 1981) qu’incombe la découverte du rôle joué par la symétrie des fonctions d’onde dans le contrôle des chemins de réaction.Il s’agit, dans le cas présent, de la symétrie spatiale des orbitales moléculaires. Woodward et Hoffmann ont considéré la réaction de fermeture de cycle de la molécule de butadiène (fig. 7). Ils ont fait remarquer que, pour une molécule portant des substituants différents X et Y aux deux bouts, deux produits distincts peuvent être envisagés. L’un est un cyclobutène, où X et Y sont du même côté du plan (cis ), et s’obtient par un mouvement concerté de rotation dans le même sens des bouts du réactif. L’autre produit, où X et Y sont de côté opposé du plan (trans ), s’obtient par un mouvement concerté en sens contraire des bouts du réactif. Les deux différents types de rotation concertée sont appelés conrotatoire et disrotatoire.En considérant la symétrie des orbitales moléculaires du réactif et du produit, Woodward et Hoffmann ont posé en principe que cette symétrie ne peut changer au cours du chemin réactionnel. On est donc obligé de faire corréler une orbitale de symétrie donnée du produit à une orbitale de même symétrie du réactif. Le «diagramme de corrélation» ainsi obtenu est différent pour le premier mouvement – où un axe de symétrie est conservé – et pour le second – où un plan de symétrie est conservé. Dans le second chemin, une orbitale basse et occupée du réactif ( 祥) corrèle avec une orbitale haute et vacante du produit. Le chemin disrotatoire est donc très coûteux en énergie et est, selon les termes de Woodward et Hoffmann, «interdit par symétrie». Expérimentalement, dans cette réaction, comme dans des centaines d’autres, le produit observé correspond bien au chemin «permis par symétrie». Il faut remarquer que, si l’un des deux derniers électrons du butadiène est excité dans l’orbitale supérieure 祥, la perte d’énergie d’un électron est compensée par le gain d’énergie de l’autre: photochimiquement, le mouvement disrotatoire devient permis et semble même plus favorable que l’autre.L’utilisation de ces diagrammes de corrélation (dits de Woodward et Hoffmann) s’est largement répandue, tant en chimie organique qu’en chimie organométallique. Cette idée de corréler la structure électronique des réactifs avec celle des produits en utilisant la symétrie comme élément de contrôle a donné naissance à plusieurs autres théories. Par exemple, il est possible de rationaliser la réactivité photochimique de nombreux composés en utilisant la symétrie de la fonction d’onde globale du système (son «état»), et non plus celle de ses orbitales moléculaires. Cette approche a été appliquée avec succès à des réactions aussi diverses que la capture d’atomes d’hydrogène ou la fragmentation des cétones. Une théorie plus récente met en jeu la corrélation des structures à liaison de valence pour les réactifs et pour les produits. Cette dernière démarche diffère aussi des deux autres par le fait qu’elle n’utilise pas la symétrie, mais la localisation spatiale des paires d’électrons. Une compréhension approfondie du mécanisme des réactions de substitution nucléophile bimoléculaires (SN2) ainsi que la rationalisation des phénomènes d’hypervalence sont deux succès à porter à l’actif de cette nouvelle théorie.
qui signifie essentiellement: «L’opération de l’opérateur hamiltonien H sur la fonction d’onde 切, fonction des coordonnées de toutes les particules (noyaux et électrons), donne la même fonction 切 multipliée par un nombre E. Le nombre E est l’énergie du système. L’équation admet généralement un ensemble de solutions E, correspondant aux différentes énergies possibles. On a donc un ensemble de niveaux d’énergie discrets pour la molécule, c’est-à-dire discontinus, ce qui est une caractéristique primordiale de la mécanique quantique (en mécanique classique, l’énergie d’un système peut varier continûment).L’opérateur hamiltonien Hop contient à la fois un terme différentiel, assimilable à l’énergie cinétique des particules, et un terme multiplicatif, assimilable à leur énergie potentielle. C’est la présence simultanée de ces deux termes de caractère mathématique très différent qui rend la résolution de l’équation si difficile.En pratique, une première simplification est apportée en séparant noyaux et électrons. Comme le noyau le plus petit, le proton, est déjà 1 860 fois plus lourd qu’un électron, on suppose que les électrons se déplacent beaucoup plus rapidement que les noyaux. Ainsi, du point de vue d’un électron en mouvement, les noyaux sont essentiellement fixes. C’est l’approximation de Born-Oppenheimer: les noyaux n’ont pas eu le temps de bouger alors que déjà les électrons ont accompli de nombreuses trajectoires.Il reste que, même réduite aux seuls électrons, l’équation de Schrödinger est impossible à résoudre analytiquement, sauf pour le cas simple d’un électron (atome hydrogène H ou ions hélium He +, lithium Li ++...). Il faut donc trouver des méthodes approchées; les deux méthodes utilisées communément correspondent chacune à un point de vue différent pour la molécule.La méthode des liaisons de valenceL’idée de la méthode des liaisons de valence est que la meilleure fonction d’onde pour une molécule est celle qui superpose les fonctions partielles, correspondant chacune à un mode satisfaisant de liaison pour la molécule. Ainsi, pour la molécule d’hydrogène H2, le chimiste peut intuitivement envisager deux modes possibles de liaison pour les deux électrons (fig. 1 a).Le premier mode correspond à un partage des deux électrons, provenant de chaque atome, entre les deux atomes, pour former une liaison «covalente». Dans le second mode, l’un des atomes accapare les deux électrons et porte ainsi une charge électrique négative; l’autre atome, dépouillé de son électron, porte une charge électrique positive. L’attraction coulombienne entre ces deux charges crée une liaison «ionique». La méthode des liaisons de valence superpose, pour construire la fonction d’onde globale 切, les fonctions d’onde correspondant à ces deux types de liaison.De la même manière, dans la molécule de benzène, les atomes de carbone – chacun pouvant former quatre liaisons avec les atomes voisins – ont le loisir d’organiser leurs liaisons de deux manières (fig. 2 a). La fonction d’onde 切 correcte est une superposition des fonctions d’onde correspondant à ces deux structures. Chimiquement, cette construction est d’autant plus satisfaisante qu’elle s’accorde avec la notion de résonance introduite par Pauling, dans laquelle la molécule de benzène est supposée en quelque sorte alterner entre ces deux structures (ou en tout état de cause emprunter ses caractéristiques chimiques simultanément à ces deux structures). On caractérise cette résonance par une flèche à double pointe.L’intérêt principal de cette méthode est de transcrire en termes quantiques le langage quotidien des chimistes qui représentent la molécule par une ou plusieurs structures de Lewis où les liaisons entre atomes proviennent de la mise en commun d’une paire d’électrons. Dans la méthode de la liaison de valence, on associe une fonction d’onde précise à chaque structure de Lewis. Malheureusement, cette simplicité conceptuelle s’accompagne de grandes difficultés numériques qui viennent seulement d’être résolues. C’est la raison pour laquelle la théorie de la liaison de valence a été nettement moins développée que la théorie des orbitales moléculaires.La méthode des orbitales moléculairesL’autre grande méthode de la chimie théorique consiste à attribuer à chaque électron de la molécule une onde (orbitale moléculaire ) recouvrant toute la molécule. Chaque molécule contient un certain nombre d’orbitales moléculaires ou ondes de type monoélectronique, obtenues comme solutions d’une équation de Schrödinger simplifiée, dite équation de Hartree-Fock. Dans cette équation, on obtient les orbitales moléculaires à l’aide d’une approche autocohérente.Ainsi, pour la molécule d’hydrogène H2, on peut construire, à partir des orbitales atomiques 1s A et 1s B des deux atomes d’hydrogène H, deux orbitales moléculaires, 祥 et 祥 (fig. 1 b). L’une de ces orbitales, 祥, a une amplitude forte entre les deux noyaux. Il lui correspond une forte densité électronique, mesurée par | 祥|2, dans la région de la liaison, ce qui tend à rapprocher les noyaux. Pour cette raison, on l’appelle orbitale moléculaire liante . L’autre orbitale moléculaire 祥 a son amplitude partagée en deux régions de signe opposé. L’un des volumes, qui symbolise l’extension de l’onde, est en gris pour indiquer le changement de signe. Sa densité dans la région internucléaire est très faible, et les noyaux tendent à se repousser; c’est une orbitale antiliante . L’orbitale liante 祥 a une énergie plus basse que celle des orbitales atomiques des atomes séparés, alors que l’orbitale antiliante a une énergie plus élevée.Dans le modèle des orbitales moléculaires, les électrons peuvent occuper par paires les orbitales de plus basse énergie. Dans ces paires, les deux électrons ont leur spin opposé, le spin étant un moment cinétique de rotation intrinsèque de l’électron, conçu comme une «toupie» autour de son axe propre. La limitation au nombre de deux électrons par orbitale est une conséquence du principe d’exclusion de Pauli, qui impose des restrictions de symétrie sur la nature de la fonction d’onde globale 切. Ainsi, les deux électrons de la molécule d’hydrogène occupent l’orbitale liante. On schématise le gain d’énergie dans la molécule par un diagramme où les électrons sont symbolisés par des flèches indiquant la direction de leur spin (fig. 1 c).Dans les hydrocarbures conjugués, comme le benzène, un rôle particulier est joué par les orbitales moléculaires 神 bâties sur les orbitales atomiques 2p z des atomes de carbone (fig. 2 b). Les paires d’électrons qui occupent des orbitales moléculaires de ce type sont délocalisées sur toute la molécule. Il y a trois paires, qui se répartissent uniformément sur les six liaisons carbone-carbone. La description orbitale moléculaire est donc intermédiaire entre les deux structures de résonance (voir méthode précédente). Dans l’une ou l’autre méthode, on aboutit correctement à une molécule de benzène symétrique avec six liaisons carbone-carbone d’égale longueur.3. Quelques découvertes importantes de la chimie théoriqueSurfaces de potentiel et «état de transition»En partant de l’approximation de Born-Oppenheimer, le chimiste théoricien peut calculer l’énergie d’une molécule pour n’importe quelle disposition relative des noyaux. Ainsi, pour une molécule diatomique, il peut évaluer l’énergie en fonction de la distance entre les deux noyaux. Il obtient une courbe appelée courbe de potentiel (fig. 3 a). Cette courbe de potentiel, comme ses formes plus compliquées appelées surfaces de potentiel lorsque le nombre d’atomes est plus grand, représente en quelque sorte la géographie «énergétique» du terrain (vallées, montagnes) que doit emprunter la molécule quand elle se déforme (fig. 3 b).Pour une molécule diatomique, la courbe de potentiel a un puits dont le fond correspond à la position d’équilibre de la molécule, entourée d’un côté par un mur brusque (noyaux trop rapprochés) et de l’autre par une montée plus douce. Cette pente douce correspond à l’élongation progressive de la liaison et à la dissociation de la molécule. Cette forme de courbe est appelée courbe de Morse .Pour réagir, une molécule doit quitter la vallée correspondant à sa géométrie initiale de réactif, pour trouver une autre vallée, correspondant à sa nouvelle forme en tant que produit de la réaction. Si plusieurs molécules entrent en réaction, la situation topologique reste la même. La ou les molécules doivent donc franchir un col pour réagir (fig. 3 c). Elles choisissent le col le plus bas qui puisse relier les deux vallées. L’existence et le rôle de ce col furent soupçonnés par Kohnstamm et Scheffer aux Pays-Bas en 1910, puis découverts et étudiés en premier par Evans et Polanyi, et séparément par Eyring en 1935. Ce dernier l’appela «état de transition». De nos jours, les théoriciens savent calculer la géométrie détaillée de ces états de transition, qui sont en fait des structures ressemblant à des molécules très déformées, où certaines liaisons sont étirées ou même coupées.Le paramagnétisme de la molécule d’oxygèneEn très grande majorité, les molécules sont neutres au point de vue magnétique. Placées dans un champ magnétique externe, elles développent seulement un petit moment magnétique induit qui s’oppose à la direction du champ. Cependant, certaines molécules se comportent comme de véritables petits aimants: elles possèdent un moment magnétique permanent. C’est le cas de la molécule d’oxygène 2: un récipient d’oxygène liquide placé en équilibre sur le plateau d’une balance fait pencher celle-ci si on approche un aimant. Cette aimantation permanente des molécules d’oxygène, dont l’effet au niveau microscopique est de renforcer le champ externe, s’appelle paramagnétisme . Un des premiers grands succès de la méthode des orbitales moléculaires a été l’explication de ce paramagnétisme.La molécule 2 contient 16 électrons, chacun des atomes d’oxygène en fournissant 8. Ces 16 électrons entrent dans les orbitales moléculaires liantes, en commençant par les plus basses. Si on laisse de côté les deux électrons très profonds de type 1s de la couche interne de chaque atome (quatre en tout), et les électrons demi-profonds de type 2s , aussi au nombre de deux par atome (quatre en tout), il reste huit électrons à placer. À partir des orbitales atomiques 2p x , 2p y , 2p z des deux atomes, on peut former trois orbitales moléculaires liantes et trois orbitales antiliantes. Six électrons entrent dans les orbitales moléculaires liantes (fig. 4). Il reste encore deux électrons; bon gré mal gré, ceux-ci entrent dans les orbitales antiliantes, dont il se trouve que les deux plus basses ont la même énergie. D’après la règle de Hund, les deux électrons doivent occuper chacun une orbitale , en gardant leurs spins dans le même sens. Comme à chaque spin d’électron est associé un petit moment magnétique, la molécule possède au total un moment magnétique correspondant à deux spins d’électron. C’est ce qui est vérifié expérimentalement.L’aromaticité et la règle de HückelLes molécules telles que benzène, naphthalène ou anthracène font partie de la famille des molécules aromatiques (cf. BENZOÏDES). Terme utilisé originellement pour caractériser l’odeur forte de ces molécules, l’«aromaticité» a depuis pris le sens d’une stabilité exceptionnelle de ces molécules hydrocarbonées cycliques ou polycycliques par rapport aux molécules analogues à chaînes ouvertes [cf. AROMATICITÉ].Cette stabilité s’explique facilement par la méthode des orbitales moléculaires. En effet, pour un anneau d’atomes de carbone, les orbitales moléculaires 神 se rangent en énergie croissante de la façon suivante: une orbitale très basse, suivie par des paires d’orbitales d’énergie semblable, enfin une orbitale très haute (fig. 5).Ainsi, pour un anneau avec 4n + 2 atomes (6, 10, 14...), il y a 2n + 1 orbitales liantes, suivies de 2n + 1 orbitales antiliantes. Les 4n + 2 électrons de type 神 remplissent de façon satisfaisante toutes les orbitales moléculaires liantes, et forment une couche «fermée» semblable à celle des atomes les plus inertes (hélium, He; néon, Ne; argon, Ar). Par contre, pour un anneau à 4n atomes (4, 8, 12...), la paire d’orbitales liantes et la paire d’orbitales antiliantes les plus centrales sont remplacées par une paire d’orbitales de caractère «non liant»: leur occupation par les électrons n’aide pas à la formation des liaisons carbone-carbone, mais ne s’y oppose pas violemment non plus. Deux électrons 神 (sur les 4n ) occupent chacun une de ces orbitales non liantes. La couche électronique est «ouverte» et la molécule beaucoup plus réactive. Les anneaux à 6, 10 ou 14 électrons doivent donc être particulièrement stables: c’est la règle de Hückel. Si l’on considère les périmètres des molécules de benzène, naphthalène et anthracène, ils ont précisément 4n + 2 atomes, et aussi 4n + 2 électrons 神. Les liaisons transannulaires du naphthalène et de l’anthracène ne modifient que très peu la grande stabilité structurelle due à ces périmètres.Structure électronique des composés inorganiquesLa théorie des orbitales moléculaires a également joué un rôle de premier plan en chimie organométallique, c’est-à-dire dans l’étude des composés possédant simultanément des atomes de métal et des molécules organiques. La structure du ferrocène, synthétisé en 1951, est un des premiers exemples dans ce domaine: la description de la liaison chimique dans cette molécule est restée longtemps obscure car il est impossible d’écrire une structure de Lewis covalente pour ce composé. La théorie des orbitales moléculaires apporte ici une description élégante de la liaison: il se forme plusieurs orbitales moléculaires liantes entre les orbitales d du métal et les orbitales moléculaires 神 de chacun des cycles pentadiényle (fig. 6). À nouveau, les électrons sont accueillis par paires dans chacune de ces orbitales moléculaires, ce qui confère une grande stabilité à cet édifice.Un autre exemple, où la théorie a joué un rôle primordial, est celui de la découverte du cyclobutadiène, anneau instable à quatre électrons. La synthèse directe de ce composé par les méthodes traditionnelles de la chimie organique est restée longtemps infructueuse. Ce sont deux théoriciens anglais, Orgel et Longuet-Higgins, qui, à partir de l’analyse de la structure électronique de ce composé, proposèrent une nouvelle voie de synthèse utilisant des dérivés métalliques. C’est effectivement par cette voie que la première synthèse du cyclobutadiène fut accomplie deux années plus tard.Enfin, la chimie théorique a aussi su expliquer, sans malheureusement avoir pu la prédire, l’existence surprenante des composés de gaz rares, tels que le difluorure ou l’hexafluorure de xénon (XeF2 ou Xe6). Elle a contribué ainsi à faire disparaître le mythe de l’inertie chimique des atomes tels que le xénon ou le krypton.Le rôle de la symétrie dans le contrôle des réactions chimiquesC’est principalement aux savants américains Woodward (prix Nobel 1965) et Hoffmann (prix Nobel 1981) ainsi qu’au chimiste japonais Fukui (prix Nobel 1981) qu’incombe la découverte du rôle joué par la symétrie des fonctions d’onde dans le contrôle des chemins de réaction.Il s’agit, dans le cas présent, de la symétrie spatiale des orbitales moléculaires. Woodward et Hoffmann ont considéré la réaction de fermeture de cycle de la molécule de butadiène (fig. 7). Ils ont fait remarquer que, pour une molécule portant des substituants différents X et Y aux deux bouts, deux produits distincts peuvent être envisagés. L’un est un cyclobutène, où X et Y sont du même côté du plan (cis ), et s’obtient par un mouvement concerté de rotation dans le même sens des bouts du réactif. L’autre produit, où X et Y sont de côté opposé du plan (trans ), s’obtient par un mouvement concerté en sens contraire des bouts du réactif. Les deux différents types de rotation concertée sont appelés conrotatoire et disrotatoire.En considérant la symétrie des orbitales moléculaires du réactif et du produit, Woodward et Hoffmann ont posé en principe que cette symétrie ne peut changer au cours du chemin réactionnel. On est donc obligé de faire corréler une orbitale de symétrie donnée du produit à une orbitale de même symétrie du réactif. Le «diagramme de corrélation» ainsi obtenu est différent pour le premier mouvement – où un axe de symétrie est conservé – et pour le second – où un plan de symétrie est conservé. Dans le second chemin, une orbitale basse et occupée du réactif ( 祥) corrèle avec une orbitale haute et vacante du produit. Le chemin disrotatoire est donc très coûteux en énergie et est, selon les termes de Woodward et Hoffmann, «interdit par symétrie». Expérimentalement, dans cette réaction, comme dans des centaines d’autres, le produit observé correspond bien au chemin «permis par symétrie». Il faut remarquer que, si l’un des deux derniers électrons du butadiène est excité dans l’orbitale supérieure 祥, la perte d’énergie d’un électron est compensée par le gain d’énergie de l’autre: photochimiquement, le mouvement disrotatoire devient permis et semble même plus favorable que l’autre.L’utilisation de ces diagrammes de corrélation (dits de Woodward et Hoffmann) s’est largement répandue, tant en chimie organique qu’en chimie organométallique. Cette idée de corréler la structure électronique des réactifs avec celle des produits en utilisant la symétrie comme élément de contrôle a donné naissance à plusieurs autres théories. Par exemple, il est possible de rationaliser la réactivité photochimique de nombreux composés en utilisant la symétrie de la fonction d’onde globale du système (son «état»), et non plus celle de ses orbitales moléculaires. Cette approche a été appliquée avec succès à des réactions aussi diverses que la capture d’atomes d’hydrogène ou la fragmentation des cétones. Une théorie plus récente met en jeu la corrélation des structures à liaison de valence pour les réactifs et pour les produits. Cette dernière démarche diffère aussi des deux autres par le fait qu’elle n’utilise pas la symétrie, mais la localisation spatiale des paires d’électrons. Une compréhension approfondie du mécanisme des réactions de substitution nucléophile bimoléculaires (SN2) ainsi que la rationalisation des phénomènes d’hypervalence sont deux succès à porter à l’actif de cette nouvelle théorie.
Encyclopédie Universelle. 2012.